nAMDを対象とした抗VEGF薬の前向き大規模臨床試験から考えるアイリーアの有効性と安全性
nAMDは慢性進行性の疾患であり、再発を繰り返すことで不可逆的な視力低下をきたすため、長期にわたり継続した治療が必要です。長期治療を行う上で重要なのは、薬剤の有効性と安全性のバランスだと考えています。治療方針について患者さんとコミュニケーションする際には、全身と眼内の安全性に関する情報を適切に把握しながら、患者さんに説明して治療を行うよう努めています。
そこで本資材では、現在までに報告されているアイリーアの有効性と安全性について、日本で承認されている他の抗VEGF薬との比較も含めた前向き大規模臨床試験を中心に紹介します。

安川 力 先生
名古屋市立大学大学院
医学研究科視覚科学(眼科)
教授
第Ⅲ相試験:VIEW1試験(海外データ)、日本人を含む第Ⅲ相国際共同試験:VIEW2試験
【アイリーアのラニビズマブに対する非劣性の検証】
本剤は海外で実施された第Ⅰ相試験、第Ⅱ相試験の結果および海外第Ⅲ相試験、日本人を含む第Ⅲ相国際共同試験を基に承認されました。承認時に評価されたデータを紹介しますが、一部国内の承認内容と異なる成績が含まれています。
試験概要
目的
中心窩下脈絡膜新生血管(CNV)を伴う滲出型加齢黄斑変性(AMD)患者を対象として、アイリーアの有効性についてラニビズマブ0.5mg4週ごと投与に対する非劣性を検証するとともに、安全性および忍容性についても検討する
試験対象
中心窩下CNVを伴う滲出型AMD患者2,457例
VIEW1試験:1,217例、VIEW2試験:1,240例(うち日本人:101例)
試験デザイン
無作為化二重遮蔽実薬対照比較試験
投与方法
対象患者を、アイリーア2mg4週ごと投与群、0.5mg4週ごと投与群、2mg8週ごと投与群、およびラニビズマブ0.5mg4週ごと投与群の4群に無作為に割り付け、硝子体内投与を行った。治療開始時は全群4週ごと投与を3回連続行い、その後は各群の投与スケジュールに従い投与した(固定投与期)。52週経過後は、各群ともに投与間隔を12週に1回を基本とし、再投与基準に合致した場合は、それ以前(4週あるいは8週)に投与できることとした(Modified Quarterly Dosing期)。
主な有効性評価項目
主要評価項目:52週目に視力が維持(ETDRS視力表による最高矯正視力文字数の低下が15文字未満)された患者の割合
副次評価項目:52週目における最高矯正視力文字数のベースラインからの変化量 など
追加評価項目:中心網膜厚(CRT)の変化量 など
探索的評価項目:96週目(2年目)終了時のすべての評価※1
主な安全性評価項目
有害事象、副作用、重篤な有害事象、投与中止に至った有害事象、死亡、APTC定義による動脈血栓塞栓事象 など
解析計画
検証的な解析
主要評価項目(PPS):アイリーア投与群のラニビズマブ投与群に対する非劣性の検証(限界値※210%)。検定の多重性を考慮し、事前に定めた順序(アイリーア2mg4週ごと投与群、アイリーア0.5mg4週ごと投与群、アイリーア2mg8週ごと投与群)に従い検定を行う。
副次評価項目(FAS):アイリーア投与群のラニビズマブ投与群に対する優越性の検証。ただし、検定の多重性を考慮し、すべてのアイリーア投与群において、主要評価項目で非劣性が検証された場合に限り、事前に定めた順序(アイリーア2mg4週ごと投与群、アイリーア0.5mg4週ごと投与群、アイリーア2mg8週ごと投与群)に従い検定を行う。
探索的な解析
追加評価項目(FAS)
探索的評価項目(FAS)
部分集団解析:VIEW2試験における日本人の部分集団解析 など
※1:VIEW2試験において、92週目に再投与が行われた患者では、100週目の結果を最終評価として用いた(CNV病変面積、NEI VFQ-25合計スコア、CRT)
※2:VIEW1試験:両側95.1%信頼区間、VIEW2試験:両側95%信頼区間
視力の維持(ETDRS視力表による最高矯正視力文字数の低下が15文字未満)
VIEW1試験、VIEW2試験ともに、52週目に視力が維持された患者の割合において、アイリーアのすべての投与群でラニビズマブに対する非劣性が検証されました。
視力が維持された患者の割合
LOCF、PPS(52週目)、FAS(96週目)

LOCF(last observation carried forward):最終評価スコア外挿法
PPS(per protocol set):治験実施計画書に適合した患者集団
FAS(full analysis set):最大の解析対象集団
52週目に視力が維持された患者の群間差
LOCF、PPS

非劣性限界値:10%
a)ラニビズマブ投与群ー各アイリーア投与群(信頼区間は正規近似を用いた)
最高矯正視力文字数のベースラインからの変化量
52週目における最高矯正視力文字数のベースラインからの変化量において、VIEW1試験では、アイリーア2mg4週ごと投与群のラニビズマブ投与群に対する優越性が検証されましたが、VIEW2試験では、アイリーア2mg4週ごと投与群のラニビズマブ投与群に対する優越性は検証されませんでした。
視力が維持された患者の割合
LOCF、FAS

CRTのベースラインからの変化量
LOCF、PPS

承認時評価資料
本邦におけるアイリーアの中心窩下脈絡膜新生血管を伴う加齢黄斑変性に対する用法及び用量
アフリベルセプト(遺伝子組換え)として2mg(0.05mL)を1ヵ月ごとに1回、連続3回(導入期)硝子体内投与する。その後の維持期においては、通常、2ヵ月ごとに1回、硝子体内投与する。なお、症状により投与間隔を適宜調節するが、1ヵ月以上あけること。
本邦におけるラニビズマブの中心窩下脈絡膜新生血管を伴う加齢黄斑変性症に対する用法及び用量
ラニビズマブ(遺伝子組換え)として0.5mg(0.05mL)を1ヵ月毎に連続3ヵ月間(導入期)硝子体内投与する。その後の維持期においては、症状により投与間隔を適宜調節するが、1ヵ月以上の間隔をあけること。
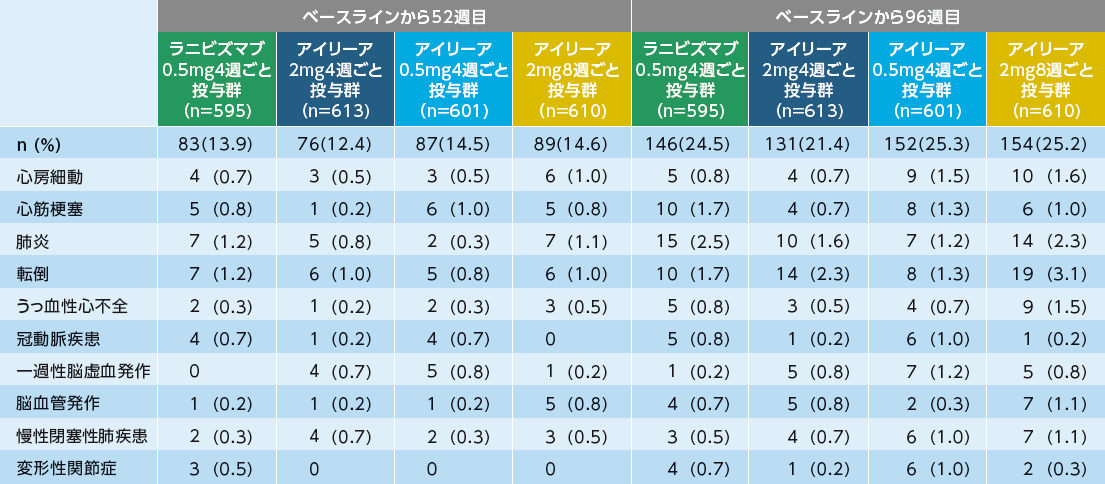
安全性
2年間の有害事象発現率
国内外で実施された第Ⅲ相試験[2試験の併合解析(2年間)]において、副作用*1は、アイリーア投与群a)に割付けられた1,824例中896例(49.1%)、ラニビズマブ投与群で595例中311例(52.3%)に認められた。
主な副作用は、アイリーア投与群で結膜出血480例(26.3%)、眼痛158例(8.7%)、眼圧上昇89例(4.9%)、ラニビズマブ投与群で結膜出血171例(28.7%)、眼痛54例(9.1%)、眼圧上昇39例(6.6%)などであった。
試験薬に関連する試験眼の重篤な有害事象
- VIEW1試験
- アイリーア投与群:1例(0.1%)[白内障]
ラニビズマブ投与群:2例(0.7%)[視力低下、偽眼内炎(各1例)]
- VIEW2試験
- アイリーア投与群:4例(0.4%)[網膜出血、視力低下、網膜色素上皮裂孔、白内障(各1例)]
ラニビズマブ投与群:1例(0.3%)[網膜色素上皮裂孔]
試験薬に関連する全身性の重篤な有害事象
- VIEW1試験
- アイリーア投与群:1例(0.1%)[脳血管発作]
ラニビズマブ投与群:0例
- VIEW2試験
- アイリーア投与群:9例(1.0%)[脳血管発作(3例)、一過性脳虚血発作、急性冠動脈症候群、ラクナ梗塞、心筋梗塞、虚血性脳卒中 、腎不全(各1例)]
ラニビズマブ投与群:0例
APTC定義による動脈血栓塞栓事象b)
- VIEW1試験
- アイリーア投与群:血管死13例(1.4%)、非致死性心筋梗塞12例(1.3%)、非致死性脳卒中8例(0.9%)
ラニビズマブ投与群:非致死性心筋梗塞8例(2.6%)、非致死性脳卒中3例(1.0%)、血管死2例(0.7%)
- VIEW2試験
- アイリーア投与群:非致死性心筋梗塞13例(1.4%)、血管死11例(1.2%)、非致死性脳卒中5例(0.5%)
ラニビズマブ投与群:非致死性心筋梗塞4例(1.4%)、非致死性脳卒中2例(0.7%)、血管死1例(0.3%)
試験薬に関連する投与中止に至った有害事象
- VIEW1試験
- アイリーア投与群:0例
ラニビズマブ投与群:偽眼内炎(1例)
- VIEW2試験
- アイリーア投与群:脳血管発作*2(3例)、黄斑変性、視力低下、網膜出血、薬疹、急性冠動脈症候群、ラクナ梗塞、虚血性脳卒中、腎不全、歩行障害*2 、会話障害*2(各1例)
ラニビズマブ投与群:0例
試験薬に関連する死亡
- VIEW1試験
- アイリーア投与群:0例
ラニビズマブ投与群:0例
- VIEW2試験
- アイリーア投与群:脳血管発作、虚血性脳卒中(各1例)
ラニビズマブ投与群:0例
発現例数(発現率%)
a) アイリーア2mg4週ごと、0.5mg4週ごと、2mg8週ごと投与群の3群合算により検討
b) すべての有害事象のうち、APTC(Antiplatelet Trialists’ Collaboration)定義により判定された動脈血栓塞栓事象
*1:投与手技に起因する有害事象を含む。添付文書に準じ、併合での集計結果を記載した。
*2:脳血管発作の1例と歩行障害および会話障害は同一症例
承認時評価資料
眼に関連する重篤な有害事象(いずれかの群で1例以上)
SAS

全身性の重篤な有害事象(いずれかの群で1例以上)
SAS
APTC定義に基づく動脈血栓塞栓事象
SAS

SAS(safety analysis set):安全性解析対象集団
Schmidt-Erfurth U, et al.: Ophthalmology. 2014; 121: 193-201
利益相反:著者のうち10名はBayerまたはRegeneronの社員である。著者にBayer、Regeneron、参天製薬よりコンサルタント料、研究費、講演料等を受領している者が含まれる。
第Ⅳ相試験:RIVAL試験(海外データ)
【ラニビズマブとアイリーアの黄斑萎縮などに関する安全性・有効性の比較】
注意:RIVAL試験はラニビズマブの第Ⅳ相試験です。
試験概要
目的
中心窩下脈絡膜新生血管を伴う加齢黄斑変性(nAMD)患者における24ヵ月にわたるTreat and Extend(T&E)投与によるラニビズマブ0.5mgとアイリーア2mgの黄斑萎縮(MA)および他の有効性の転帰の違いを検討する
試験対象
未治療のnAMD患者278例(50歳以上、ベースライン時のLogMAR視力表による最高矯正視力文字数≧23文字)
試験デザイン
多施設共同無作為化部分遮蔽第Ⅳ相臨床比較試験
投与方法
対象患者をラニビズマブ0.5mg投与群、アイリーア2mg投与群に1:1に無作為に割り付け、各薬剤を導入期投与としてベースライン、4週後、8週後に投与後、両群とも同じT&Eレジメン(最小投与間隔4週、最大投与間隔12週とし、2週幅で延長を行う)に従って硝子体内投与した。短縮する場合は、次の基準に従って短縮幅を決定した。[①最高矯正視力文字数から5文字以上の視力低下、②新規の網膜出血の存在、③光干渉断層計(OCT)上の網膜内液または網膜下液の存在、の①~③のうち、1つ該当した場合は2週短縮、2つ以上該当した場合は4週間隔まで短縮。]
主な安全性評価項目
主要評価項目:ベースラインから24ヵ月後のMAの平方根面積の変化
重要な副次評価項目:ベースラインから12ヵ月後の抗VEGF薬の投与回数 など
副次評価項目:ベースラインから12ヵ月後のMAの平方根面積の変化、ベースラインから24ヵ月後の抗VEGF薬の投与回数 など
安全性評価項目:すべての来院時における眼および全身性の有害事象(重篤な有害事象、投与中止に至った有害事象、死亡 など)、網膜神経線維(RNF)に関する分析、ベースラインおよび3回目投与の7日後(9週時)における眼の炎症
解析計画
主要評価項目(FAS)、副次評価項目(FAS):<ベースラインから12ヵ月後、24ヵ月後のMAの平方根面積の変化>変量効果混合モデルを使用し、共変量の1つとしてベースラインのMA面積を含めた。モデルには応答変数としてベースラインから12ヵ月後および24ヵ月後のMA面積の変化量を、固定効果として連続変数のベースラインMA面積、治療、来院日および治療と来院日の相互作用を含めた。小病変の拡大率の遅さを補正するため平方根変換を使用した。患者は変量効果としてモデリングした。12ヵ月後および24ヵ月後に最小二乗平均値とその95%信頼区間を治療群ごとに推定し、群間差、95%信頼区間およびp値も12ヵ月目および24ヵ月目に推定した。
重要な副次評価項目(FAS)、副次評価項目(FAS):<ベースラインから12ヵ月後、24ヵ月後の抗VEGF薬の投与回数>
安全性評価項目(SAS):眼および全身性の有害事象および眼の炎症は記述統計を使用して要約した。RNFに関する分析にはベースラインおよび24ヵ月後の平均RNF層(RNFL)厚を使用した。
MAの平方根面積※のベースラインからの変化
※:MAを正方形にした場合の1辺の長さ
ベースラインから24ヵ月後のMAの平方根面積の変化について、治療群間に有意差は認められませんでした。
FAS

最小二乗平均値±標準偏差
抗VEGF薬の投与回数
FAS

平均値±標準偏差
安全性
SAS
有害事象※はラニビズマブ0.5mg投与群で141例中125例(88.7%)、アイリーア2mg投与群で139例中130例(93.5%)に認められました。
※:治療薬との因果関係は問わない
眼に関連する主な有害事象(いずれかの群で2例以上)

死亡および重篤な有害事象

N/A =データ不明。
有害事象の報告にはMedDRA version17が用いられた。
†:ラニビズマブ0.5mg投与群(心不全、大腿骨頸部骨折、転移性食道がん 各1例)、アイリーア2mg投与群(急性腎不全、副腎がん、血管免疫芽球性T細胞リンパ腫、心不全、閉塞性尿路疾患を伴う悪性新生物の進行、代謝性アシドーシス 各1例)
試験薬に関連する死亡は認められなかった。
有害事象による投与中止
ラニビズマブ0.5mg投与群:141例中9例
アイリーア2mg投与群:139例中14例
重篤な有害事象による投与中止
ラニビズマブ0.5mg投与群:141例中7例
アイリーア2mg投与群:139例中14例
RNFL厚の減少(ベースラインから10μm超)
<12ヵ月後>
ラニビズマブ0.5mg投与群:122例中3例(2.5%)
アイリーア2mg投与群:111例中3例(2.7%)
<24ヵ月後>
ラニビズマブ0.5mg投与群:110例中4例(3.6%)
アイリーア2mg投与群:97例中1例(1%)
眼の炎症(9週まで)
<グレード1+の前房内細胞>
ラニビズマブ0.5mg投与群:約5%
アイリーア2mg投与群:約7%
<グレード1の前房フレア>
ラニビズマブ0.5mg投与群:約6%
アイリーア2mg投与群:約7%
Gillies MC, et al.: Ophthalmology. 2020; 127: 198-210
利益相反:著者にBayerより謝礼、研究費等を受領している者、Bayerのアドバイザリーボードメンバー等が含まれる。
日本人を含む国際共同第Ⅲ相試験:HAWK試験、第Ⅲ相試験:HARRIER試験(海外データ)
【ブロルシズマブのアイリーアに対する非劣性の検証】
注意:HAWK試験、HARRIER試験はブロルシズマブの第Ⅲ相試験です。
HAWK試験にはブロルシズマブ3mg投与群が含まれていますが、国内の承認用法及び用量の範囲を逸脱しているため、承認の範囲内の症例群のみに限定し、一部改変しています。
試験概要
目的
48週の最高矯正視力のベースラインからの変化量に関して、ブロルシズマブ6mgのアイリーア2mgに対する非劣性を検証する。
試験対象
50歳以上で治療対象眼が未治療の活動性CNVを伴う滲出型加齢黄斑変性患者
HAWK試験:1,078例(うち日本人患者は154例)、HARRIER試験:739例
試験デザイン
多施設共同二重遮蔽無作為化並行群間比較試験
投与方法
対象患者をブロルシズマブ3mg投与群(HAWK試験のみ)、ブロルシズマブ6mg投与群、アイリーア2mg投与群のいずれかに1:1:1で無作為に割り付け、導入期投与として各薬剤を4週ごとに3回硝子体内投与した後、維持期投与としてブロルシズマブ3mg投与群または6mg投与群は12週ごと、アイリーア2mg投与群は8週ごとに硝子体内投与した(16週目の評価まではブロルシズマブ6mg投与群とアイリーア2mg投与群の治療曝露期間が同一)。ブロルシズマブ3mg投与群または6mg投与群では、規定来院日に遮蔽医師による疾患活動性評価を行い、疾患活動性が認められた場合は試験終了時まで8週ごとに投与した。
主要評価項目
最高矯正視力文字数※のベースラインからの変化量(48週)
最も重要な副次評価項目
最高矯正視力文字数※のベースラインからの平均変化量(36~48週)
その他の副次評価項目
最高矯正視力文字数※のベースラインからの変化量(96週、各評価時点)
中心サブフィールド厚(CST)のベースラインからの変化量(16週、48週、36~48週および96週、各評価時点) など
安全性評価項目
有害事象(治療対象眼および眼以外) など
解析計画
主要評価項目、最も重要な副次評価項目
FAS(無作為化された患者のうち、治験薬が1回以上投与された患者)を解析対象集団とし、欠測値はLOCF法を用いて補完した。ベースライン後の測定値がない患者ではベースライン値を、治験薬投与中止後も試験を継続した患者では治験薬以外のVEGF阻害剤を使用した時点で治療対象眼での有効性評価打切りとして、打切り直前のデータを用いた。
解析は、群、ベースラインの最高矯正視力区分(≦55文字、56~70文字、≧71文字)、年齢区分(<75歳、≧75歳)を固定効果とした分散分析(ANOVA)を行い、投与群間差(ブロルシズマブ6mg投与群-アイリーア2mg投与群)の両側95%信頼区間の下限が非劣性マージンの-4文字より大きい場合に、アイリーア2mg投与群に対するブロルシズマブ6mg投与群の非劣性が検証されることとした(有意水準は片側0.025)。
多重性の調整は階層的手法を適用し、主要評価項目、最も重要な副次評価項目の順にそれぞれブロルシズマブ6mg投与群 vs. アイリーア2mg投与群、ブロルシズマブ3mg投与群 vs. アイリーア2mg投与群の順に、先行する評価項目について非劣性が検証された場合に次の評価項目の非劣性を検証できることとした。
その他の副次評価項目
FASを解析対象集団とし、各評価時点の最高矯正視力スコアのベースラインからの変化量および各評価時点のCSTのベースラインからの変化量については、主要評価項目と同様に解析した。HAWK試験では、主要評価項目および最も重要な副次評価項目で非劣性が検証された場合には、CSTのベースラインからの変化量に対する優越性検定をブロルシズマブ6mg投与群、ブロルシズマブ3mg投与群の順に実施することとし、16週、48週、36~48週の評価項目ごとに階層的にブロルシズマブ6mg投与群またはブロルシズマブ3mg投与群とアイリーア2mg投与群を比較して、先行する評価項目について優越性が確認された場合に次の検定を実施できることとした(CSTのベースラインからの変化量の有意水準は片側0.005)。
なお、ブロルシズマブ6mg投与群は、12週ごとに投与した患者と8週ごとに投与した患者を併合解析した。
※:ETDRS文字数(ETDRS視力検査表を用いて測定開始距離4mで評価)
最高矯正視力文字数のベースラインからの変化量1,2)
(主要評価項目:48週、最も重要な副次評価項目:36~48週、その他の副次評価項目:96週、各評価時点)
LOCF、FAS
HAWK試験、HARRIER試験ともに、48週における最高矯正視力文字数のベースラインからの変化量、および36~48週における平均変化量において、ブロルシズマブ6mg投与群のアイリーア2mg投与群に対する非劣性が検証されました。
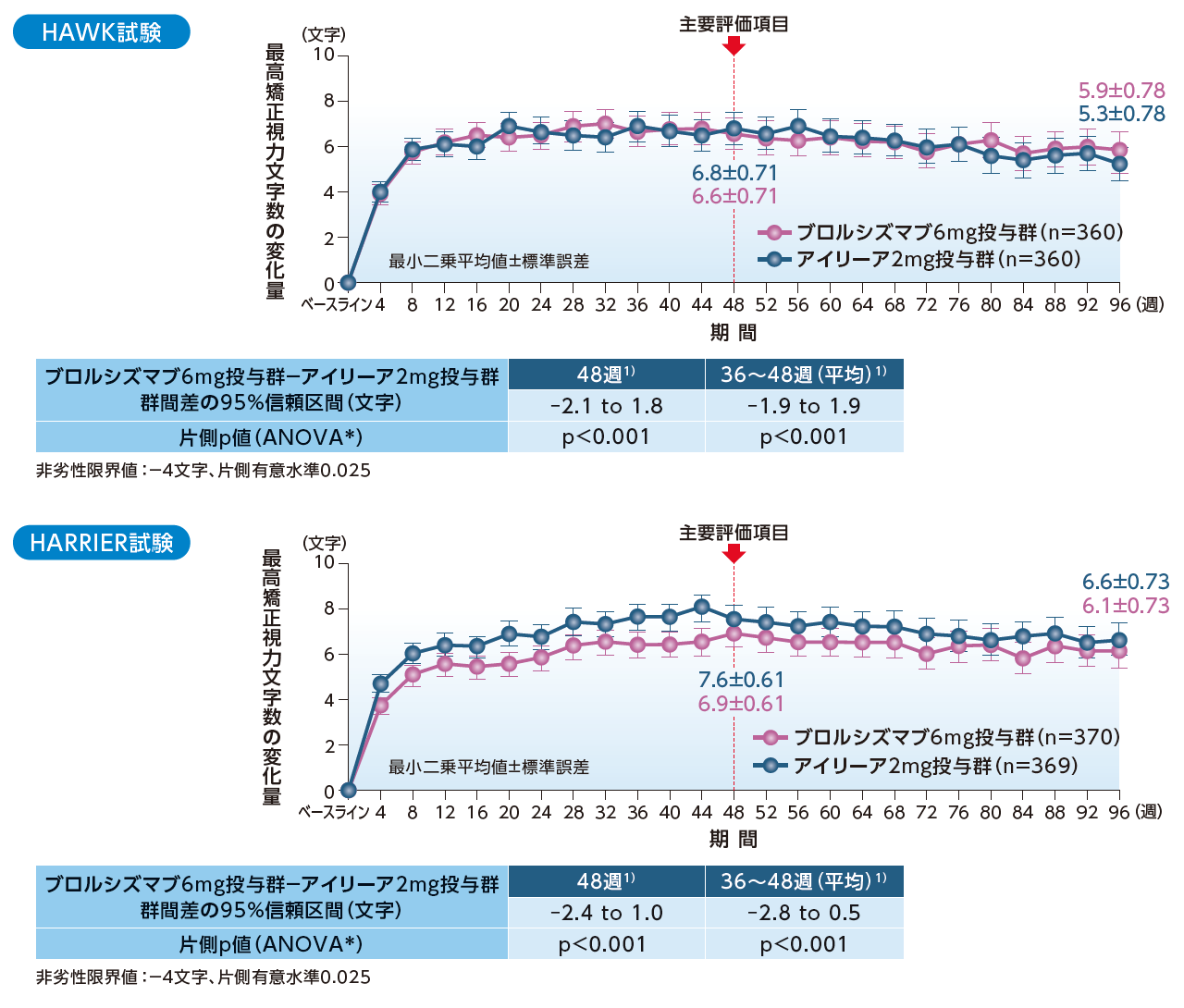
*:ベースラインの最高矯正視力区分(≦55文字、56~70文字、≧71文字)、年齢区分(<75歳、≧75歳)、群を固定効果とした
CSTのベースラインからの変化量1,2)
(その他の副次評価項目:16週、48週、36~48週および96週、各評価時点)
LOCF、FAS
HAWK試験では、16週および48週のCSTのベースラインからの変化量において、ブロルシズマブ6mg投与群のアイリーア2mg投与群に対する優越性が検証され、36~48週のCSTのベースラインからの平均変化量において、優越性は検証されませんでした。

*:ベースラインのCST区分(<400μm、≧400μm)、年齢区分(<75歳、≧75歳)、群を固定効果とした
- 1)
- Dugel PU, et al.: Ophthalmology. 2020; 127: 72-84
利益相反:本論文の著者に参天製薬のサイエンスアドバイザリーボードのメンバー、Bayer、参天製薬のコンサルタント、Bayer、参天製薬、Regeneronより謝礼、助成金、講演料を受領している者が含まれる。
- 2)
- Dugel PU, et al.: Ophthalmology. 2021; 128: 89-99
利益相反:本論文の著者に参天製薬のサイエンスアドバイザリーボードのメンバー、Bayer、参天製薬、Regeneronのコンサルタント、Bayer、参天製薬、Regeneronより経済的支援、Bayerより講演料を受領している者が含まれる。
安全性
有害事象[96週]
眼に関連する主な有害事象(治療対象眼、いずれかの群で2%以上)
眼に関連する主な有害事象は、HAWK試験においてブロルシズマブ6mg投与群で360例中220例(61.1%)、アイリーア2mg投与群で360例中201例(55.8%)、HARRIER試験においてブロルシズマブ6mg投与群で370例中174例(47.0%)、アイリーア2mg投与群で369例中176例(47.7%)に認められた。

眼に関連する重篤な有害事象(SAE)(治療対象眼)
眼に関連するSAEは、HAWK試験においてブロルシズマブ6mg投与群で360例中12例(3.3%)、アイリーア2mg投与群で360例中5例(1.4%)、HARRIER試験においてブロルシズマブ6mg投与群で370例中13例(3.5%)、アイリーア2mg投与群で369例中6例(1.6%)に認められた。
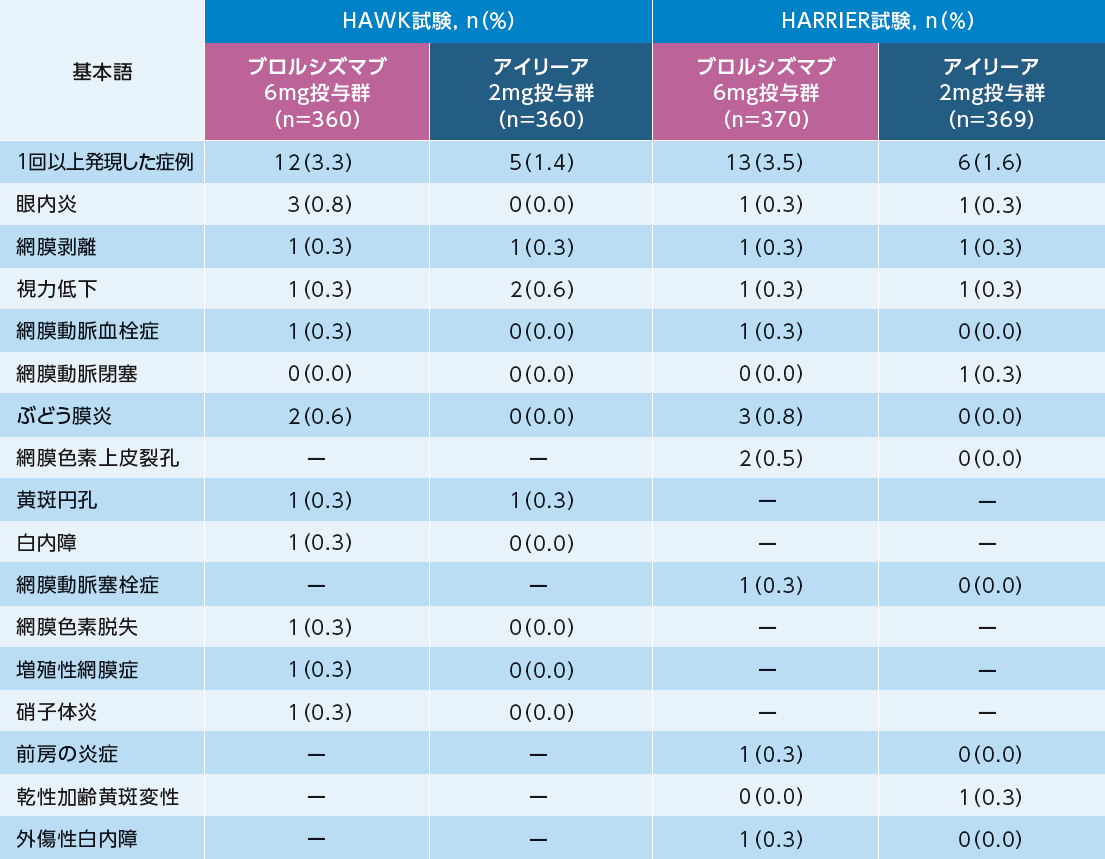
-:本試験で報告されていない
眼以外の重篤な有害事象※
●HAWK試験
ブロルシズマブ6mg投与群で10例、アイリーア2mg投与群で9例に肺炎が認められた。
●HARRIER試験
ブロルシズマブ6mg投与群で下肢骨折が3例、失神が3例、アイリーア2mg投与群で肺炎が8例に認められた。
※:論文には最も多く報告された有害事象のみ掲載されている
Dugel PU, et al.: Ophthalmology. 2021; 128: 89-99
日本人を含む国際共同第Ⅲ相試験:TENAYA試験、第Ⅲ相試験:LUCERNE試験(海外データ)
【ファリシマブのアイリーアに対する非劣性の検証】
注意:TENAYA試験、LUCERNE試験はファリシマブの第Ⅲ相試験です。
試験概要
目的
中心窩下脈絡膜新生血管を伴う加齢黄斑変性(nAMD)患者を対象に、ファリシマブを最長16週まで延長して投与したときの有効性についてアイリーア8週ごと投与に対する非劣性を検証するとともに、安全性及び忍容性についても検討する。
試験対象
50歳以上の未治療のnAMD患者
TENAYA試験:671例、LUCERNE試験:658例
試験デザイン
多施設共同二重遮蔽無作為化並行群間比較試験
投与方法
対象患者をファリシマブ6mg投与群とアイリーア2mg投与群に1:1で無作為に割り付け、ファリシマブ6mg投与群は、導入期投与としてファリシマブ6mgを4週ごとに4回硝子体内投与した後、20週及び24週時の疾患活動性評価に基づき投与間隔を8週、12週または16週のいずれかで60週時まで固定投与し、その後は108週時までPTIレジメン※1で継続した。アイリーア2mg投与群は、導入期投与としてアイリーア2mgを4週ごとに3回硝子体内投与した後、108週時まで8週ごとに投与した。
主要評価項目
40、44、48週時の最高矯正視力文字数※2のベースラインからの変化量平均値
副次評価項目
最高矯正視力文字数のベースラインからの変化量の推移
40、44、48週時の中心サブフィールド厚(CST)のベースラインからの変化量平均値およびベースラインからの変化量の推移 など
安全性評価項目
有害事象(治療対象眼および眼以外) など
解析計画
ITT(無作為化された全ての患者)を有効性解析対象集団とし、欠測データはMMRMにより暗黙的に補完した。
主要評価項目の解析は、応答変数として4~48週時の最高矯正視力文字数のベースラインからの変化量、固定効果として、カテゴリ共変量である投与群、時点、投与群と時点の交互作用、ベースラインの最高矯正視力文字数、ベースラインの最高矯正視力文字数(≦54文字、55~73文字、≧74文字)、低輝度条件下での視力低下(<33文字、≧33文字)、地域を含めたMMRMモデルを用い、投与群間差(ファリシマブ6mg投与群とアイリーア2mg投与群との差)の95.03%信頼区間の下限が非劣性マージンの-4文字より大きい場合に、アイリーア2mg投与群に対するファリシマブ6mg投与群の非劣性が検証されることとした。
副次評価項目は、主要評価項目と同様に解析した。
※1:PTI(personalized treatment interval)レジメンでは、薬剤投与来院時のCST、最高矯正視力文字数及び臨床評価に基づいて投与間隔を調整(8週、12週または16週)した。
※2:ETDRS文字数(EDTRS視力検査表を用いて測定開始距離4mで評価)
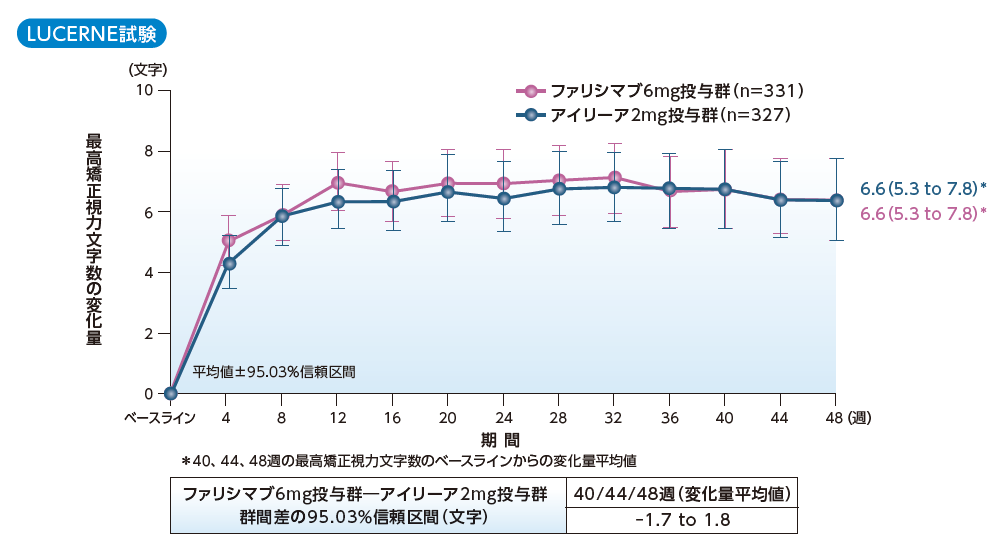
最高矯正視力文字数のベースラインからの変化量
(主要評価項目:40/44/48週、副次評価項目:変化量の推移)
MMRM、ITT
TENAYA試験、LUCERNE試験ともに、40、44、48週時の最高矯正視力文字数のベースラインからの変化量平均値において、ファリシマブ6mg投与群のアイリーア2mg投与群に対する非劣性が検証されました。

CSTのベースラインからの変化量(副次評価項目:40/44/48週、変化量の推移)
MMRM、ITT

*40、44、48週時点のCSTのベースラインからの変化量平均値
ファリシマブ6mg投与群-アイリーア2mg投与群の群間差(95.03%信頼区間):-7.4(-15.7, 0.8)

*40、44、48週時点のCSTのベースラインからの変化量平均値
ファリシマブ6mg投与群-アイリーア2mg投与群の群間差(95.03%信頼区間):-6.4(-14.8, 2.1)
Heier JS, et al.: Lancet. 2022; 399(10326): 729-740.
利益相反:本論文の著者にBayer、Regeneronよりコンサルタント料、助成金、講演料を受領しているものが含まれる。
安全性
SAS
有害事象[48週]
眼に関連する主な有害事象(治療対象眼、いずれかの群で2%以上)
眼に関連する主な有害事象は、TENAYA試験においてファリシマブ6mg投与群で333例中121例(36.3%)、アイリーア2mg投与群で336例中128例(38.1%)、LUCERNE試験においてファリシマブ6mg投与群で331例中133例(40.2%)、アイリーア2mg投与群で326例中118例(36.2%)に認められた。

眼以外の主な有害事象(いずれかの群で2%以上)
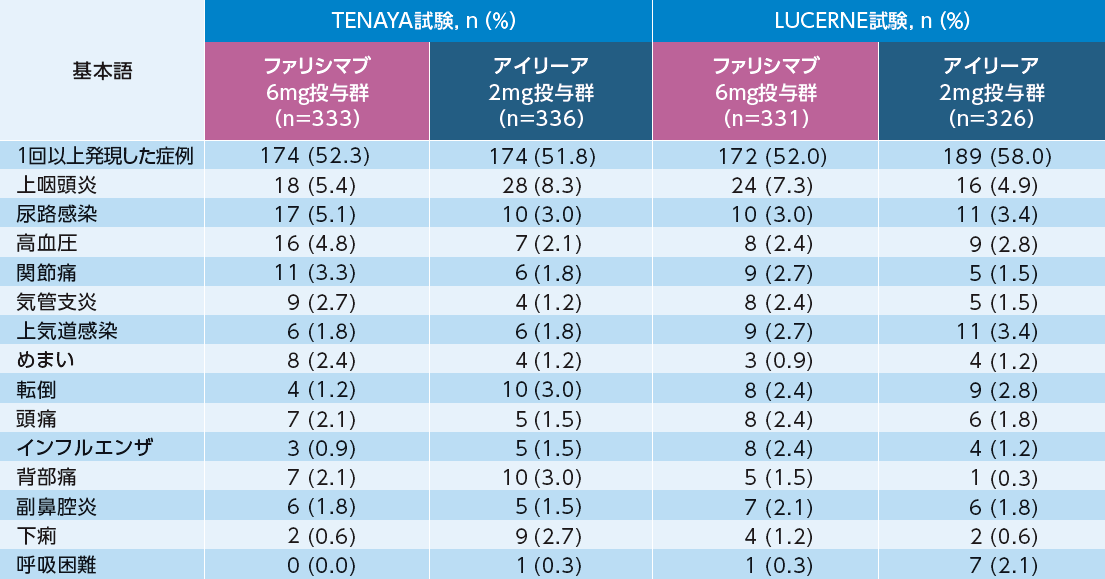
眼以外の主な有害事象は、TENAYA試験においてファリシマブ6mg投与群で333例中174例(52.3%)、アイリーア2mg投与群で336例中174例(51.8%)、LUCERNE試験においてファリシマブ6mg投与群で331例中172例(52.0%)、アイリーア2mg投与群で326例中189例(58.0%)に認められた。
眼に関連する重篤な有害事象(SAE)(治療対象眼)
眼以外の主な有害事象は、TENAYA試験においてファリシマブ6mg投与群で333例中174例(52.3%)、アイリーア2mg投与群で336例中174例(51.8%)、LUCERNE試験においてファリシマブ6mg投与群で331例中172例(52.0%)、アイリーア2mg投与群で326例中189例(58.0%)に認められた。

眼以外の重篤な有害事象(SAE)※
●TENAYA試験
ファリシマブ6mg投与群で333例中30例、アイリーア2mg投与群で336例中34例に認められた。
●LUCERNE試験
ファリシマブ6mg投与群で331例中38例、アイリーア2mg投与群で326例中48例に認められた。
※論文には発現例数と発現率のみ掲載されている。
有害事象による投与中止※
●TENAYA試験
ファリシマブ6mg投与群で3例3件(眼障害1件[nAMDの悪化]、筋骨格系および結合組織障害、神経系障害各1件)、アイリーア2mg投与群で3例3件(良性/悪性および詳細不明の新生物3件)であった。
●LUCERNE試験
ファリシマブ6mg投与群で8例8件(眼障害5件[虹彩毛様体炎、網膜色素上皮裂孔、ぶどう膜炎、硝子体炎、硝子体剥離]、神経系障害2件、心臓障害1件)、アイリーア2mg投与群で1例1件(眼障害1件[ぶどう膜炎])であった。
※論文には眼障害以外の投与中止に至った有害事象は器官別大分類のみ記載されている。
APTC定義により判定された動脈血栓塞栓事象に基づく死亡※
●TENAYA試験
ファリシマブ6mg投与群:2例
アイリーア2mg投与群:1例
●LUCERNE試験
ファリシマブ6mg投与群:0例
アイリーア2mg投与群:2例
※論文にはAPTC(Antiplatelet Trialists’Collaboration)定義により判定された動脈血栓塞栓事象に基づく死亡のみ記載されている。
Heier JS, et al.: Lancet. 2022; 399(10326): 729-740.
利益相反:本論文の著者にBayer、Regeneronよりコンサルタント料、助成金、講演料を受領しているものが含まれる。
